What are Langmuir Films?
Langmuir Films are thin organic films of a thickness of just one molecule and are the source of high expectations. As useful components in many practical and commercial applications such as sensors, detectors, displays and electronic circuit components. With both the possibility to synthesize custom organic molecules and sophisticated thin film deposition technology it is possible to create electrically, optically and biologically active components on a nanometer scale.
A thin organic film can be deposited on a solid substrate by various techniques. These include thermal evaporation, sputtering, electrodeposition, molecular beam epitaxy, adsorption from solution, Langmuir-Blodgett technique, and self-assembly. The Langmuir- Blodgett (LB) technique is one of the most promising methods for the preparation of thin films as it enables
(A) precise control of the monolayer thickness,
(B) homogeneous deposition of the monolayer over large areas and
(C) the possibility to make multilayer structures with varying layer composition.
An additional advantage of the LB technique is that monolayers can be deposited on almost any kind of solid substrate
Some terms to help understand Monolayers
Surface Tension
Surface tension is a measurement of the cohesive energy present at an interface. All of the molecules of a liquid attract each other. These interactions of molecules in the bulk of a liquid are balanced so that equal forces attract them in all directions. Molecules on the surface of a liquid, however, experience an imbalance of forces as indicated below.
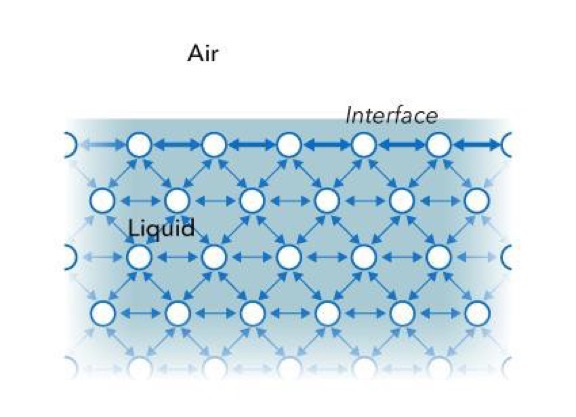
The excess energy present at the surface is called surface free energy and can be quantified as a measurement of energy/area. It is also possible to describe this situation as having a line tension or a surface tension which is quantified as a force/length measurement. The most common units for surface tension are dynes/cm or mN/m. These units are equivalent.
The surface is the interface of two fluids. The experiment is referred to as a surface tension measurement if it involves a gaseous phase. If two liquids are tested then the experiment is referred to as an interfacial tension measurement. In either case, the denser fluid is referred to as the heavy phase and the less dense fluid is referred to as the light phase. Solids also may be described to have a surface free energy at their interfaces but direct measurement of its value is not possible through the techniques used for liquids.
Polar liquids like water have strong intermolecular interactions and thus have high surface tension. Any factor which decreases the strength of the interaction will lower the surface tension, and so an increase to the temperature of the system will lower surface tension. Any contamination, especially by surfactants, will lower surface tension which is why researchers should be very concerned about cleanliness and preventing contaminants!
Surfactants
Surfactants, or surface-active agents, are a larger class of molecules which have a significant technological and biological importance. Generally, these molecules consist of a hydrophilic (water soluble) and a hydrophobic (water insoluble) component. This amphiphilic (consists of both hydrophilic and hydrophobic components) nature of surfactants is responsible for their behavior in a solution with the forming of micelles, bilayers, vesicles, etc. and their accumulation at interfaces (air/water or oil/water). The hydrophobic part usually consists of hydrocarbon or fluorocarbon chains, while the hydrophilic part consists of a polar group (-OH, -COOH, -NH, -PO4(CH2)2NH3, etc.).
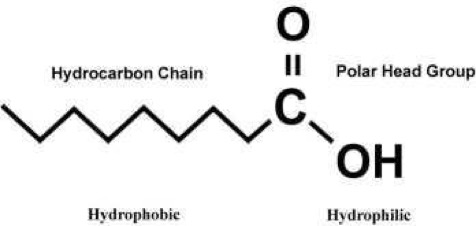
The association behavior of surfactants in solution and their affinity for interfaces is determined by the physical and chemical properties of the hydrophobic and hydrophilic groups, respectively. The shape and size of the hydrocarbon moiety and the size, charge, and hydration of the hydrophilic head group are of utmost importance in this respect. Depending on the balance between these properties a wide variety of self-assembled structures, both at interfaces and in bulk, have been observed. The driving force behind the association is the reduction of the free energy of the system. Therefore, when a surfactant comes in contact with water it accumulates at the air/water interface causing a decrease in the surface tension of water.
Insoluble monolayers
There exists a wide range of surfactants with an amphiphilic nature which drastically lowers the surface tension of water. Many of these amphiphilic substances insoluble in water can, with the help of a volatile and water-insoluble solvent, be easily spread on a water surface to form an insoluble monolayer at the air/water interface. These monolayers, also called Langmuir (L) films, represent the extreme case when considering adsorption to interfaces because all molecules are concentrated in a one molecule thick layer at the interface. The amphiphilic nature of the surfactants dictates the orientation of the molecules at the interface (air/water or oil/water) in such a way that the polar head group is immersed in the water and that the long hydrocarbon chain is pointing towards air, gas or oil.
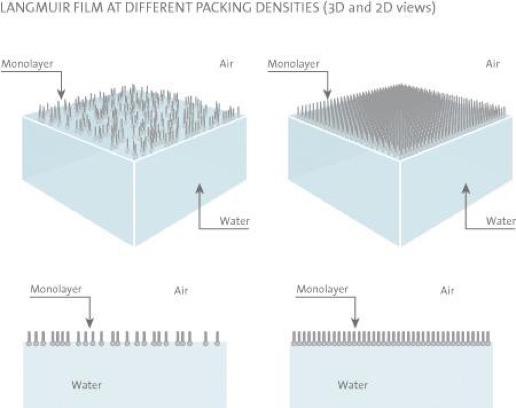
The hydrocarbon chain of the substance used for monolayer studies has to be long enough in order to be able to form an insoluble monolayer. A rule of thumb is that there should be more than 12 hydrocarbons or groups in the chain ((CH2)n, n > 12). If the chain is shorter, though still insoluble in water, the amphiphile on the water surface tends to form micelles. These micelles are water soluble, which prevents the build-up of a monolayer at the interface. On the other hand, if the length of the chain is too long the amphiphile tends to crystallize on the water surface and consequently does not form a monolayer. It is difficult to determine the optimal length for the hydrocarbon chain because its film-forming ability also depends on the polar part of the amphiphile. Furthermore, the amphiphile has to be soluble in some organic solvent which is highly volatile and water-insoluble (to transport the surfactant to the surface, chloroform or hexane is commonly used).
Surface Pressure
As was mentioned earlier, the air/water interface possesses excess free energy originating from the difference in environment between the surface molecules and those in the bulk. This interfacial free energy is accessible by measurements of the surface tension ∂. The surface tension of water is around 72 mN/m at 20°C which is an exceptionally high value compared to other liquids and consequently makes water a very good subphase for monolayer studies.
The most common way to transport an amphiphile to the surface is to dissolve it in a volatile non-polar (water-insoluble) solvent and then using a microsyringe to deposit onto the surface. The solution spreads rapidly to cover the available area. As the solvent evaporates, a monolayer is formed. When the available area for the monolayer is large the distance between adjacent molecules is large and their interactions are weak. The monolayer can then be regarded as a two-dimensional gas. Under these conditions, the monolayer has little effect on the surface tension of water. If the available surface area of the monolayer is reduced by a barrier system (see below), the molecules start to exert a repulsive force on each other.
This two-dimensional analog of pressure is called surface pressure, π, and is given by the relationship:
π = γ0 – γ
Equation 1
γ0 = surface tension of subphase without a monolayer γ = surface tension of subphase with a monolayer present
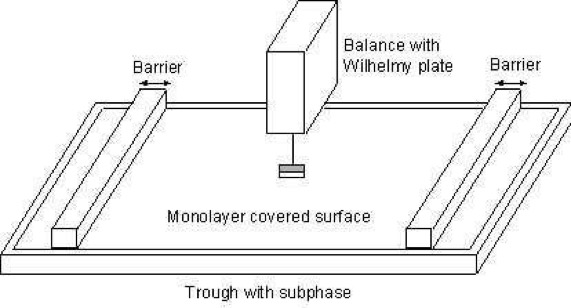
The Langmuir Film Balance
The trough holding the subphase is usually made of Teflon to prevent any leakage of the subphase over the edges. The trough is thermostatted by circulating water in channels placed underneath the Teflon trough. The surface area of the trough can be varied by sweeping movable barriers over the surface of the trough. The barriers are made of Delrin, a hydrophilic material, and heavy enough to prevent any leakage of the monolayer beneath the barrier. The surface pressure and the mean molecular area are continuously monitored during the compression.
The mean molecular area can be determined by monitoring the distance the barriers have moved and with the known dimensions of the trough. The surface pressure is measured by the Wilhelmy plate method. In this method, a measurement is made by determining the force due to surface tension on a plate suspended so that it is partially immersed in the subphase (see below). This force is then converted into surface tension (mN/m or dynes/cm) with the help of the dimensions of the plate.
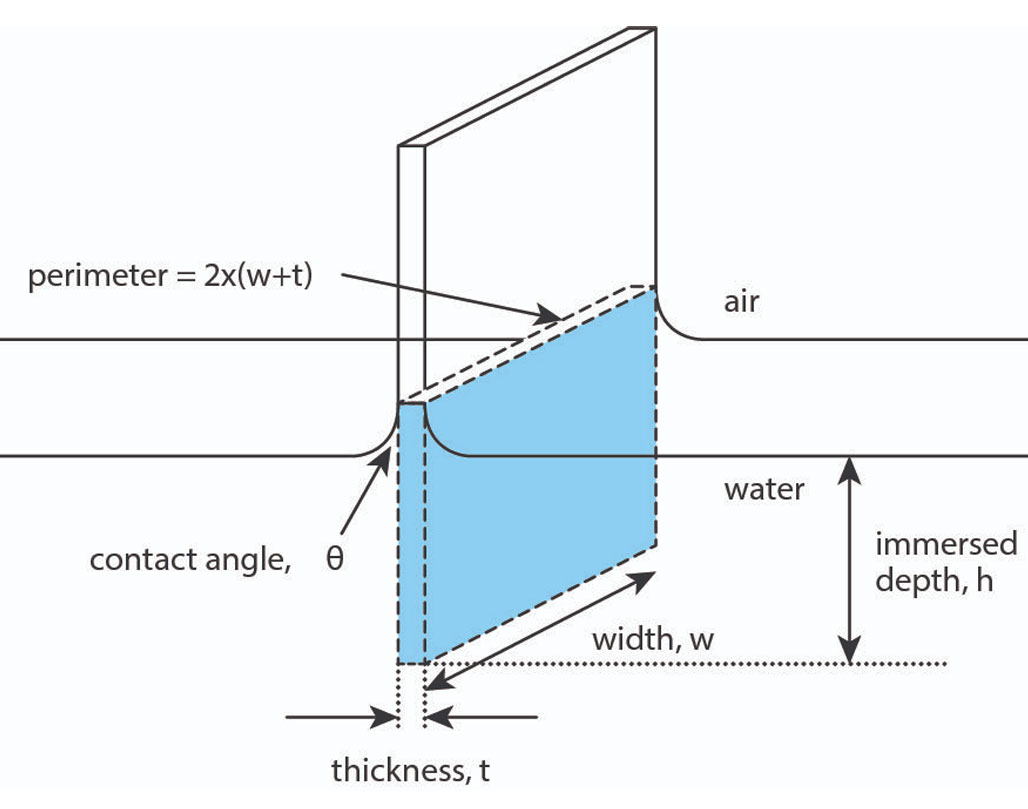
The plate is often very thin and made of platinum, but even plates made of glass, quartz, mica and filter paper can be used. The forces acting on the plate consist of the gravity and surface tension downward, and buoyancy due to displaced water upward. For a rectangular plate of dimensions lp, wp and tp, of material density ρp, immersed to a depth hl in a liquid of density ρl, the net downward force is given by the following equation:
F = ρpglpwptp + 2γ(tp + wp)(Cosθ) – ρlgtlwlhl
Equation 2
Where ρ is material density, g is gravity, l is length, w is the width, t thickness, γ is surface tension
The surface pressure is then determined by measuring the change in F for a stationary plate between a clean surface and the same surface with a monolayer present. If the plate is completely wetted by the liquid (i.e. cosθ = 1) the surface pressure is then obtained from the following equation:
π = -Δγ = -[ΔF / 2 (tp+wp)] = -ΔF/2wp, if wp >> tp
Equation 3
Where ρ is material density, g is gravity, l is length, w is the width, t thickness, γ is surface tension
The sensitivity can thus be increased by using a very thin plate. The force is in this way determined by measuring the changes in the mass of the plate, which is directly coupled to a sensitive electrobalance. The monolayer can also be held at a constant surface pressure, which is a computer-enabled controlled feedback system between the electrobalance and the motor responsible for the movements of the compressing barrier. This is useful when producing LB films i.e. when the monolayer is deposited on a solid substrate.
There are even other ways to control the area of the monolayer and to measure the surface pressure but the constructions above are the most commonly used.
Surface Pressure Against Mean Molecular Area Isotherms
The most important indicator of the monolayer properties of an amphiphilic material is given by measuring the surface pressure as a function of the area of water surface available to each molecule. This is carried out at constant temperature and is known as a surface pressure – area isotherm or simply isotherm. Usually, an isotherm is recorded by compressing the film (reducing the area with the barriers) at a constant rate while continuously monitoring the surface pressure. Depending on the material being studied, repeated compressions and expansions could be necessary to achieve a reproducible trace. A schematic p-A-isotherm is shown below.
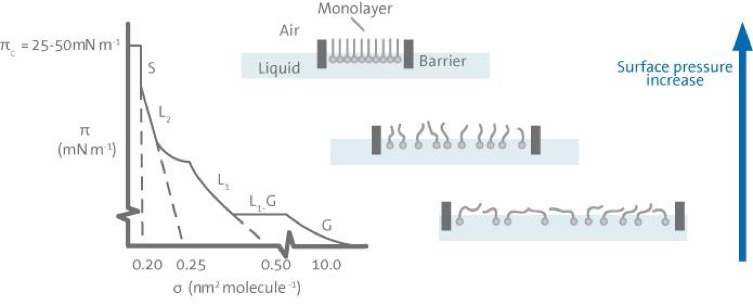
A number of distinct regions are immediately apparent in examining the isotherm. These regions are called phases. As one can see when the monolayer is compressed it can pass through several different phases which are identified as discontinuities in the isotherm. The phase behavior of the monolayer is mainly determined by the physical and chemical properties of the amphiphile, the subphase temperature, and the subphase composition. For example, various monolayer states exist depending on the length of the hydrocarbon chain and the magnitude of other cohesive and repulsive forces existing between head groups. An increase in the chain length increases the attraction between molecules, condensing the p-A-isotherm. On the other hand, if an ionizable amphiphile is used the ionization of the head groups induces repulsive forces tending to oppose phase transitions.
A simple terminology used to classify different monolayer phases of fatty acids has been proposed by W. D. Harkins as early as 1952. At large the monolayers exist in the gaseous state (G) and can on compression undergo a phase transition to the liquid- expanded state (L1). Upon further compression, the L1 phase undergoes a transition to the liquid-condensed state (L2), and at even higher densities the monolayer finally reaches the solid state (S). If the monolayer is further compressed after reaching the S state the monolayer will collapse into three-dimensional structures. The collapse is generally seen as a rapid decrease in the surface pressure or as a horizontal break in the isotherm if the monolayer is in a liquid state.
There are also many other critical points in a p-A-isotherm such as the molecular area at which an initial, pronounced increase in the surface pressure is observed, Ai, and the surface pressures at which phase transitions occur between the L1 and L2 state and the L2 and S state.
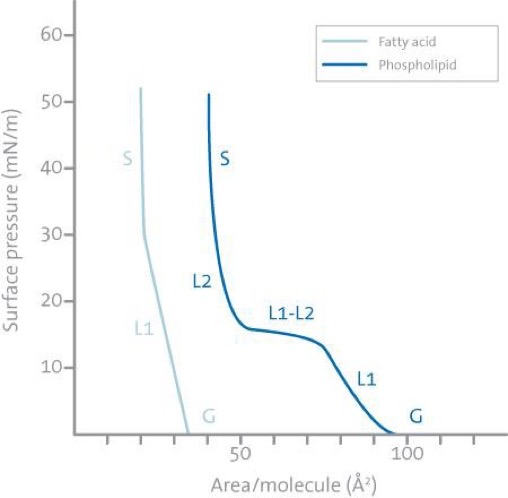
Typical isotherms of a fatty acid with a single hydrocarbon chain (left) and a phospholipid with two hydrocarbon chains (right) are illustrated in the image below. Following the definitions above one can see that the fatty acid has three distinct regions gas (G), liquid (L1) and solid (S), while the phospholipid has an additional almost horizontal transition phase (L2-L1) between the two different liquid phases. This is very common for phospholipids and the position of this horizontal transition phase is very temperature dependent. As the temperature is increased, the surface pressure value at which the horizontal transition phase occurs will increase and vice versa.
Deposition
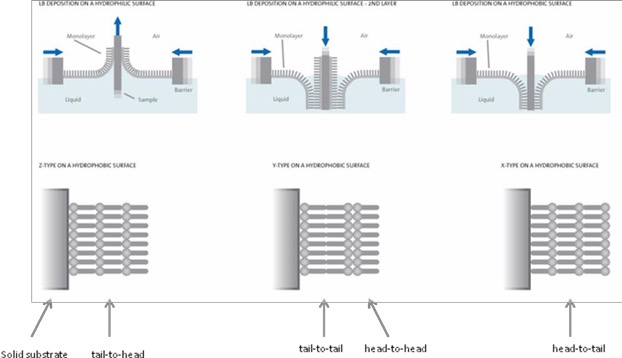
Apart from being used as a tool for monolayer studies, the Langmuir film balance can also be used for building up highly organized multilayers of an amphiphile. This is accomplished by successively dipping a solid substrate up and down through the monolayer while simultaneously keeping the surface pressure constant with a computer-controlled feedback system between the electrobalance measuring the surface pressure and the barrier moving mechanism. Consequently, the floating monolayer is adsorbed to the solid substrate. In this way, multilayer structures of hundreds of layers can be produced. These multilayer structures are commonly called Langmuir-Blodgett (or simply LB) films. The deposition process is shown below.
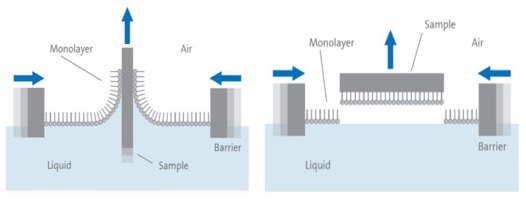
LB deposition is traditionally carried out with the monolayer in the solid phase. The surface pressure is then high enough to ensure sufficient cohesion in the monolayer, e.g. the attraction between the molecules in the monolayer is high enough so that the monolayer does not fall apart during transfer to the solid substrate. This also ensures the build-up of homogeneous multilayers. When the surface pressure is too high the increase in surface pressure caused by the solid substrate can be enough to collapse the monolayer.
The surface pressure value that gives the best results depends on the nature of the monolayer and is usually established empirically. Amphiphiles can seldom be successfully deposited at surface pressures lower than 10 mN/m, and at surface pressures above 40 mN/m collapsing and film rigidity often poses problems.
When the solid substrate is hydrophilic (glass, SiO2 etc.) the first layer is deposited by raising the solid substrate from the subphase through the monolayer, whereas if the solid substrate is hydrophobic (HOPG, salinized SiO2 etc.) the first layer is deposited by lowering the substrate into the subphase through the monolayer.
There are several parameters that affect what type of LB film is produced. These are the nature of the spread film, the subphase composition, and temperature, the surface pressure during the deposition and the deposition speed, the type and nature of the solid substrate and the time the solid substrate is stored in air or in the subphase between the deposition cycles.
The quantity and the quality of the deposited monolayer on a solid support are measured by a so-called transfer ratio, t.r. This is defined as the ratio between the decrease in monolayer area during a deposition stroke, Al, and the area of the substrate, As. For ideal transfer, the t.r. is equal to 1. Depending on the behavior of the molecule the solid substrate can be dipped through the film until the desired thickness of the film is achieved. Different kind of LB multilayers can be produced and/or obtained by successive deposition of monolayers on the same substrate (see above). The most common one is the Y-type multilayer, which is produced when the monolayer deposits to the solid substrate in both up and down directions. When the monolayer deposits only in the up or down direction the multilayer structure is called either Z-type or X-type. Intermediate structures are sometimes observed for some LB multilayers and they are often referred to as XY-type multilayers.
The production of so-called alternating layers which consist of two different kinds of amphiphiles is also possible by using highly sophisticated instruments. In such an instrument, there is a trough with two separate compartments both possessing a floating monolayer of a different amphiphile. These monolayers can then be alternatingly deposited on one solid substrate.
Another way to deposit the monolayer is the Langmuir-Schaefer (LS) technique. This technique differs from the vertical technique described above only in the sense that the solid substrate is horizontally lowered in contact with the monolayer.
References
Adamson, A. W., Physical Chemistry of Surfaces, Wiley&Sons (1976)
Wu, S., Polymer Interface & Adhesion, Marcel Dekker, N.Y. (1982)
Couper, A., Investigations of Surfaces and Interfaces – Part A, B.
Rossiter, R. C. Baetzold eds, Phys. Meth. Chem Ser vol XI A (1993)
Hansen, F. K., and Rodsrun, G., J. Coll & Inter Sci 141, 1-12(1991)
Roberts, G., Ed. Langmuir-Blodgett Films, Plenum Press, New York (1990)
Swalen, J.D.; Allara, D.L.; Andrade, J.D.; Chandross, E.A.; Garoff, S.; Israelachvili, J.; McCarthy, T.J.; Murray, R.; Pease, R.F.; Rabolt, J.F.; Wynne, K.J.; Yu, H. Langmuir, 3 (1987) 932
Breton, M. J. Macromol. Sci. Rev. Macromol. Chem., C21 (1981) 61
Petty, M.C. Thin Solid Films, 210/211 (1992) 417
Pockels, A. Nature, 46 (1892) 418
Pockels, A. Nature, 43 (1891) 437
Langmuir, I. J. Am. Chem. Soc., 39 (1917) 1848
Blodgett, K.B. J. Am. Chem. Soc., 57 (1935) 1007
A.W.Adamson, Physical Chemistry of Surfaces, Wiley & Sons, New York (1976)
Shaw, D.J., Introduction to Colloid and Surface Chemistry, Butterworth & Co, London (1980)
Laughlin, R.G. The Aqueous Phase Behaviour of Surfactants, Academic Press Inc., San Diego (1994)
Chattoraj, D.K.; Birdi, K.S. Adsorption and the Gibbs Surface Excess, Plenum Press, New York (1984)
Gaines, G.L., Insoluble Monolayers at the Liquid-Gas Interface, Wiley-Interscience, New York (1966)
Binks, B.P. Adv. Colloid Interface Sci., 34 (1991) 343
Harkins, W.D., The Physical Chemistry of Surface Films, Reinhold, New York (1952)